
Steven G. Ayre MD
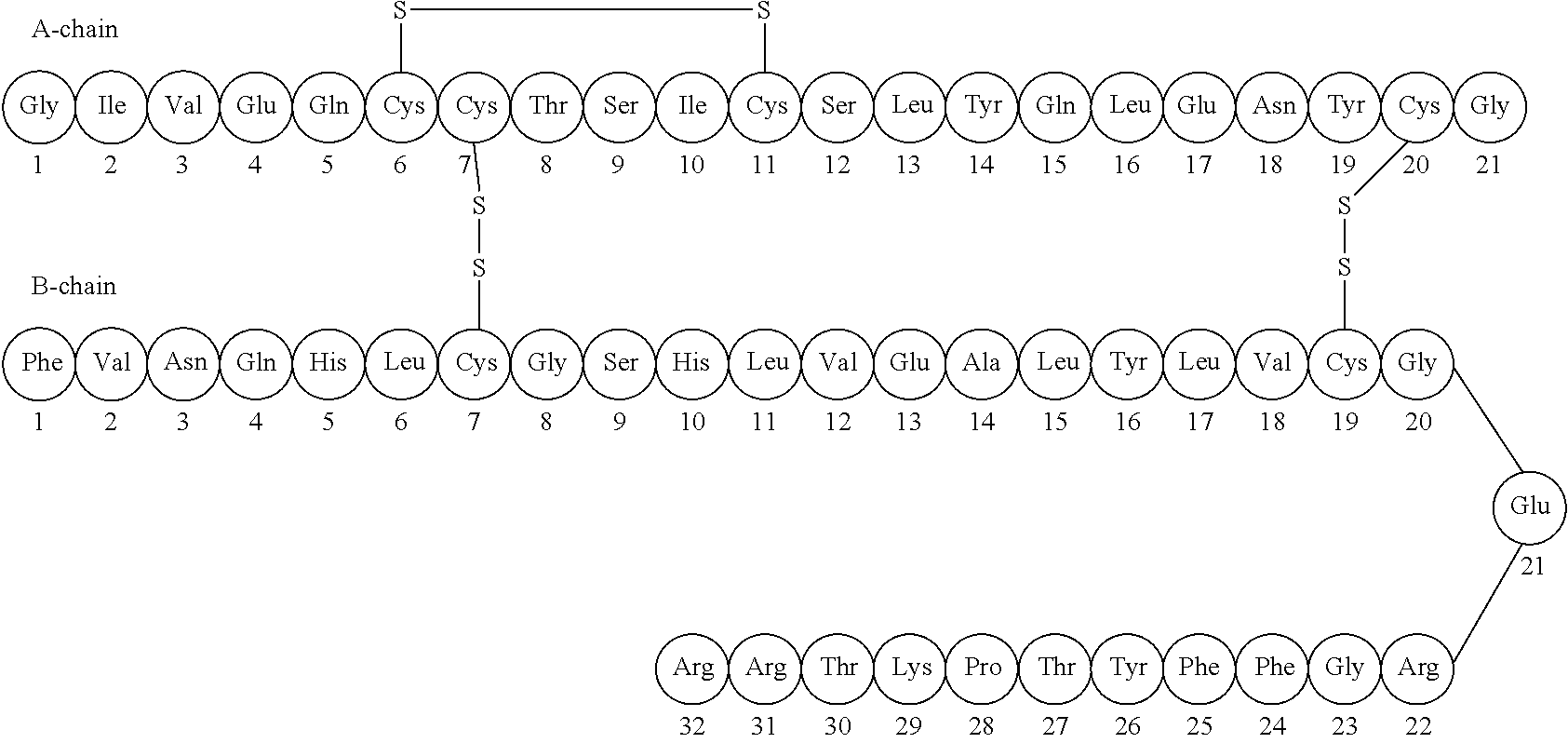
The hormone insulin is recognized as having actions that affect the transmembrane transport of different substances, particularly glucose, into numerous different kinds of cells. Insulin is a large polypeptide molecule with a molecular weight of 5808. It consists of an A chain and a B chain, connected together by two disulfide bridges. (fig 1) The hormone is made in the beta cells of the pancreas, and the stimulus for its secretion into the blood stream is a rise in the blood glucose concentration. Its actions on liver, adipose tissue, and skeletal muscle have all been studied in great detail, and it is now recognized that insulin also affects a wide variety of tissues in addition to just these three.(1)
Apart from the membrane transport of glucose, insulin also regulates the transport of some amino acids, some fatty acids, potassium, magnesium, and certain other monosaccharides. Furthermore, it mediates the formation of macromolecules in cells which are used in cell structure, energy stores, and in the regulation of many cell functions. It stimulates glycogenolysis, lipogenesis, proteogenesis, and nucleic acid synthesis. It also increases glucose oxidation and magnesium-activated sodium-potassium ATPase activity.(1)
There is a single mechanism involved in the initiation of all these biological effects, and this is the interaction of the hormone with its specific receptor. The insulin receptor consists of two alpha subunits (Mr 135,000) and two beta subunits (Mr 95,000) which are linked together by disulfide bonds. The alpha unit is predominantly located on the outer surface of the cell membrane, and the insulin binding domain is located here. The transmembrane beta subunit contains tyrosine kinase activity on its cytoplasmic domain that results in rapid receptor autophosphorylation. Activation of the kinase towards exogenous substrates is apparently preceded by this insulin-dependent autophosphorylation reaction of the beta subunit. Action on other cellular substrates ultimately leads to the expression of the full range of insulin actions at the cellular level.(2)
After insulin binds to the receptor with activation of the kinase, followed by receptor autophosphorylation, the insulin-receptor complex is endocytosed into the cell cytoplasm. This phenomenon accounts for the down-regulation of insulin receptor activity that ensues following insulin stimulation. With this endocytosis, a variety of events may then take place. Insulin dissociates from the receptor and, following fusion of the endocytotic vesicle with cellular lysosomes, it is degraded by lysosomal enzymes. The free receptor may itself be degraded by the lysosomal enzymes, or it may recycle back to the surface of the cell membrane. Finally, the free phosphorylated receptor may proceed to activate other substrates in the cytoplasm or within cellular organelles (Golgi apparatus, nucleus, etc) to produce the plethora of changes described above.(3)
The most commonly recognized action of insulin is that of lowering blood glucose. This is accomplished via a process of facilitated diffusion across cell membranes. It is hypothesized that the mechanism of this facilitated diffusion involves the translocation of a glucose transport protein from the cytoplasm out to the cell membrane. This translocation process involves the fusion of intracytoplasmic vesicles with the membrane of the cell. These vesicles contain the glucose transport protein in their enclosing membranes. Once exteriorized on the cell surface, the transport proteins serve as channels for glucose to enter the cell. This particular protein has been identified as a 40,000 molecular weight moiety found by centrifugation to be associated with the Golgi rich fraction.(4) The process of translocation is reversible via endocytosis of the membrane fragment containing the transport proteins, reconstituting the intracytoplasmic vesicles. The whole activity of the glucose transport protein is dependent on metabolic energy, and independent of protein synthesis.(5) The precise nature of the signal through which insulin turns this process on and off remains to be elucidated.
Insulin receptors are widely distributed in mammalian organisms with there being from 100 to 100,000 receptors per cell in different tissues. Rarely are there any cells having no receptors at all.(6) A number of malignant neoplastic tissues have also been found to have a plentiful supply of insulin receptors,(7-9) reflecting established cancer cell metabolism and the need that malignant cells have for glucose. Insulin may also play a role here in the stimulation of cancer cell growth,(10,11) and many different cancers have been found to actually produce and secrete their own insulin.(12-19) The conclusion to be made here is that insulin receptors on cancer cell membranes, plus autocrine secretion of insulin by cancer cells, function as an endogenous mechanism evolved in these cells allowing them to parasitize host energy substrate (glucose), and to stimulate their rapid and autonomous growth.
Investigation of many of the actions of insulin on insulin receptors in numerous species have demonstrated that the properties of insulin receptors in mammalian tissues are remarkably similar, irrespective of cell type.(1,6,20) This being so, it may be anticipated that what the activated insulin/insulin-receptor complex does in one tissue, it will do in all. This would of course be dependent on there being the necessary metabolic machinery within a particular tissue to react to insulin activation. Not all tissues are similarly endowed in this regard.
Brain is a tissue which does have insulin receptors, but which does not have the same insulin-dependent glucose transport mechanism common to many other of the body’s tissues. Insulin receptors are found both on the capillary endothelium of the BBB, as well as on the glial elements within the substance of the brain. These receptors do not seem to play any role, in conjunction with insulin, in the transmembrane transport of the glucose which is so essential for proper brain metabolism. The capillary endothelium of the blood-brain barrier (BBB) has its own unique transport system for glucose, as well as a number of other nutrient transport systems for substances such as choline, adenine, adenosine, lactate, glutamate, phenylalanine, and arginine.(21) The composition of the scant interstitial fluid of the brain is carefully controlled by the very selective functioning of the BBB. Having access to this space, across the BBB, substances then have free access to the brain cells.
The glucose transport system in brain responds to chronic changes in blood glucose levels, and there is some interesting clinical correlation for this. The system is up-regulated during prolonged periods of hypoglycemia(22) which can explain why some patients with chronic hypoglycemia or insulinomas may not have symptoms of brain glucopenia at blood glucose concentrations of less than 50 mg%. In a similar fashion, the brain glucose transport system is down-regulated during prolonged periods of hyperglycemia, such as can occur with poorly controlled diabetes.(23) When such patients are brought under rapid control with insulin therapy, because of this down-regulation of the BBB glucose transporter, they may develop symptoms of hypoglycemia due to CNS glucopenia even though the blood glucose level may be in the normal range.(24)
Glucose transport across the BBB is insulin-independent, and yet insulin receptors are found on the same BBB capillary endothelium which carries the glucose transport system. This insulin transport system is just one of a number of peptide transport systems found on the BBB. Others carry the insulin-like growth factors I and II, and transferrin.(21) The blood-brain barrier insulin receptor is a glycoprotein having structural characteristics typical of the insulin receptor in peripheral tissues. It may be part of a combined endocytosis-exocytosis (transcytosis) system for the transport of the peptide through the BBB in man. A transcytosis of insulin through the human BBB would allow for distribution of circulating insulin into brain interstitial space and insulin action on brain cells.(25)
The role of insulin in the regulation of brain function continues to be a major unsolved problem in insulin physiology. Evidence to date shows that it seems to be primarily involved with brain growth and development, and this seems to be more important in the newborn mammalian brain.(26) Research continues in efforts to elucidate this question in its entirety. It has been through such research, looking to find the extent of insulin’s role in brain physiology, that some interesting possibilities have come to light concerning the applications as a pharmacologic adjunct which this hormone may have in clinical situations other than in the management of diabetes mellitus.
In tissues possessing insulin receptors, including the membranes of the capillary endothelial cells comprising the BBB, it seems that insulin can potentiate the pharmacologic actions of drugs that may be administered in conjunction with it. In an experiment measuring the brain-uptake index in rats there was seen to be a 33 percent increase in the intra-CNS accumulation of radiolabeled AZT in the insulin pretreated animals as compared to non-insulin treated controls.(27) Drug potentiation appears to be a function of increased intracellular concentration of drug obtained due to some action of insulin on the target cell membranes. The question as to the exact mechanism of insulin’s pharmacologic action in this non-diabetic context remains an open one. Research in the field points to several different possibilities.
In skeletal muscle, insulin has been shown to deliver enzyme-insulin-albumin conjugates into the intracellular compartment of the cells. The whole complex was transported into the cells by a process resembling receptor-mediated endocytosis, and the enzyme-albumin-insulin complex retained its enzymatic activity and its ability to bind antibodies to insulin.(28) In experiments with rat fibroblasts, the fragment A of diphtheria toxin conjugated to insulin gained access to the intracellular milieu via a process of endocytosis through insulin receptors,(29) and in human lymphocytes, insulin has been shown to carry a photoactivatable psoralen derivative into these cells, again by a process of insulin receptor-mediated endocytosis.(30)
In brain, it has been pointed out that specific peptide receptor transport systems in the blood-brain barrier may be available for peptide delivery into the brain, and it has been suggested that coupling peptides or even enzymes to insulin could result in the uptake of the chimeric peptide by cells via the insulin receptor-mediated uptake system.(31) This concept has been investigated in an animal experiment with rats, wherein it was shown there is a statistically significant increase in the brain-uptake index of 3H-zydovudine under the influence of insulin.(32) In this case, it was free drug that was co-administered along with insulin, and not a chimeric drug-or-enzyme/ insulin complex. No determination has been made as to whether or not this observed effect was due to an insulin receptor-mediated phenomenon as in the cases cited above. Other research indicates that there may be alternative possibilities here.
Breast and colon cancer cell membranes have been characterized as having plentiful insulin receptors.(7-9) Autoradiographic studies have shown that radiolabeled insulin binds predominantly to breast cancer cell membranes rather than to stromal elements (fat cells, firbrolasts) within tumors.(7) Other studies have demonstrated that, quantitatively, there are six times more insulin receptors on breast cancer cell membranes than on membranes of stromal cells within tumors.(33, 34) And most significantly for the purposes of this discussion, yet another study demonstrated that, in vitro, insulin increased the cytotoxic effect of methotrexate in MCF-7 human breast cancer cells by a factor of up to ten thousand.(35) The authors of this study attributed this effect to metabolic modification within the cancer cells, rendering them more sensitive to the effects of the methotrexate. However, in a related study it was shown that “insulin has significant effects on the intramembrane methotrexate transport system of MCF-7 (human breast cancer) cells. Enhanced cytotoxicity may be related to an increased capacity of the cells to accumulate free intracellular methotrexate. Insulin-induced changes in cellular lipid synthesis and perhaps in membrane lipid profile could result in changes in membrane fluidity and enhanced methotrexate transport.”(36)
In another research context unrelated to the actions of insulin, experiments manipulating the chemical structure and physical properties of membrane phospholipids has made it possible to alter phase transitions of fluidity in the membranes that come to incorporate these compounds, and to thereby influence and control biological membrane processes. Alkyl glycerides have been shown to modify the properties of biological membranes quickly and reversibly to increase the permeation of active compounds. An important example of this is the improved transport of cytostatic drugs across the blood-brain barrier in the presence of l-pentylglycerol.(37)
Insulin is recognized as having a widespread effect on lipid metabolism, and the following may explain its putative drug-potentiating effect. It is recognized that cell membrane permeability varies directly with cell membrane fluidity, and the fluidity of cell membranes is a function of the degree of unsaturation of its component fatty acids on account of the lower melting point of unsaturated versus saturated fatty acids. Insulin has a particularly significant effect on the activity of the enzyme delta-9 desaturase, which catalyses the transformation of the saturated fatty acid stearic acid into the mono-unsaturated oleic acid.(38) The melting point of the triacylglycerol, tristearin, (with three stearic acid residues attached to a glycerol backbone) is 73° C, while that of the corresponding trioleic congener is only 5.5° C. At physiologic temperatures, a widespread transformation of this sort would account for considerable changes in the physical properties of biomembranes, and would significantly affect cell membrane permeability.(39)
In summary, though not yet definitively characterized as to its specific mechanisms, there is compelling evidence upon which to propose the following. Through its interaction with specific insulin receptors widely distributed in human malignant tissues, insulin facilitates the passage of drug molecules from the extracellular compartment into the intracellular compartment of these cells. Rather than relying just on the law of mass action in concert with relatively high doses of parenteral anticancer drugs, insulin used as a pharmacologic adjunct to lowered dose therapy accords a potentially safer as well as more effective methodology.
Insulin and a related compound – IGF-I – are integral parts the mechanisms of malignancy in cancer cells. The combination of insulin and IGF-I operates autonomously at the cellular level within the tumor, and this operation is free from any higher level of integrated control. The two work together in an autocrine and/or paracrine manner and in a complementary fashion, with IGF-I being the major anabolic hormone responsible for mediating messages about growth in the tumor, while insulin regulates and provides the fuel for these processes.(40)
An added dimension to insulin’s drug potentiation in malignant neoplastic tissues is its effect on cancer cell growth via cross-reaction with the IGF-I receptor. It is well recognized that the cell-cycle phase-specific anticancer drugs work best on cells in S-phase of the growth cycle. Growth in cancer cells is mediated by a number of different mitogens, one of the most potent of which in breast cancer cells is insulin-like growth-factor I(IGF-I).(33,41,42) IGF-I – like insulin – is manufactured and secreted by many cancer cell lines, and cancer cell membranes are – again, as for insulin – liberally endowed with the specific receptors for this mitogen.(41,42) Furthermore, there is 45 percent homology between the amino acid sequence of the insulin receptor (IR) and the insulin-like growth-factor I receptor (IGF-IR), and both insulin and IGF-I can effectively cross-react with both of these receptors.(43) As stated above, human cancer cell membranes, particularly breast, have been characterized as possessing far more IR and IGF-IR than the cell membranes of normal tissues within the host and, finally, it is a well recognized fact of mammalian physiology that the intensity of a ligand’s effect on a tissue is a function of the specific receptor concentration on that target tissue.
Thus, overall, the role of insulin in Insulin Potentiation Therapy is to stimulate cancer cell membrane insulin receptors to facilitate drug entry into cells, and to cross-react with cancer cell membrane IGF-I receptors causing a recruitment of the cancer cells into S-phase, making them more susceptible to the pharmacologic action of anticancer medication. The synergy between these actions of insulin potentiates drug effects within the cancer cells, resulting in a more effective cell kill. Furthermore, because of the much richer distribution of insulin and IGF-I receptors on cancer cell membranes versus normal somatic cells, these drug potentiating effects will predominate in the cancer cells with a relative sparing of normal tissues.
INSULIN-INDUCED HYPOGLYCEMIA IN INSULIN POTENTIATION THERAPY
The dosage of insulin used in applications of insulin potentiation therapy (IPT) is 0.1 to 0.4 U/kg body weight, given as a single bolus injection intravenously. The 0.4U/kg dose is the most widely used because it provides optimal hypoglycemic responses, is well tolerated clinically, and is easily controlled medically. The lower doses are used for individuals exhibiting increased sensitivity to insulin as evidenced by greater than the desired levels of hypoglycemia. The type of insulin used is Humalog, insulin lispro injection, recombinant DNA origin (Lilly). This is the preferred preparation because it is reported to have a faster onset of action, as well as a shorter overall duration of action of only two hours. The clinical experience with Humalog bears out the fact of its shorter duration of action, however the onset of the desired level of hypoglycemia which it produces has been observed to be no faster than that observed with Humulin or other regular insulin preparations of animal origin.
Regular insulin may be administered in other non-diabetic circumstances in clinical medicine. Growth hormone secretion in humans is evaluated by means of the insulin tolerance test, wherein subjects receive 0.05 to 0.1 U/kg body weight.(44) Also, the evaluation of insulin resistance in subjects with Syndrome-X employs a continuous infusion of low-dose insulin over a number of hours. It is not possible to compare doses administered to these Syndrome-X subjects with the dose given in IPT because of the differences in the pharmacodynamics and physiologic response seen with a bolus dose versus the continuous, long term infusion.
On the other hand, it is quite evident that the dose of insulin used in IPT is significantly higher than that given in the insulin tolerance test. While the degree of hypoglycemia produced in IPT is therefore more pronounced, this may be satisfactorily managed through close clinical observation of the subject over time followed, when appropriate, by the administration of hypertonic glucose solution. Competent and experienced management of insulin-induced hypoglycemia during treatment is central to the safe performance of the IPT protocol.
A number of studies have been done in human subjects to determine the factors important in glucose counterregulation following insulin-induced hypoglycemia.(45-48) In these studies, it has been shown that insulin-induced hypoglycemia stimulates the release of glucagon, epinephrine, growth hormone, cortisol, and norepinephrine (from sympathetic nerves). Glucagon plays the primary role in restoration of normoglycemia, while neither growth hormone, cortisol nor norepinephrine contribute to immediate glucose homeostasis in these test situations. Epinephrine plays a secondary role in the process, and this is only important when glucagon secretion is deficient.
Patients with longstanding type I diabetes mellitus have a deficient glucagon response to insulin-induced hypoglycemia, and hypoglycemic responses to insulin would be particularly profound in those patients who may also be also taking beta blocker medication (diminished epinephrine response). While growth hormone and cortisol secretion do not play a role in the immediate recovery from insulin-induced hypoglycemia, patients with hypofunction in their secretion of cortisol (Addison’s Disease, Sheehan’s Syndrome) must be handled with greater care. These patients, as well as those with type I diabetes mellitus, and those receiving beta blocker therapy should be started with the 0.1U/kg dose, and observed carefully. As IPT therapy is usually done in a series of twice-weekly or weekly treatments, these subjects may have their insulin dose gradually increased in increments of 0.05U/kg at successive treatments, as tolerated.
The clinical experience with the IPT protocol has demonstrated that hypoglycemic symptoms have their onset approximately 25 to 30 minutes after the insulin administration. This timing corresponds to the nadir glucose concentration measured in the aforementioned studies. The actual timing by the clock of the onset of hypoglycemia is not as reliable as is clinical observation of hypoglycemic symptoms. Subjects characteristically experience adrenergic symptomatology at this time (sweating, tachycardia) and this is when a 25 cc quantity of 50 percent hypertonic glucose solution is administered – directly after administration of whatever drug therapy as indicated by the diagnosed condition being treated.
The administration of 50 percent hypertonic glucose has consistently proven effective in supporting the body’s physiological responses geared to reestablish normoglycemia (glucagon immediately, growth hormone and cortisol in the longer term). It has been consistently observed to be adequate to serve to avoid any of the more profound (neuroglycopenic) symptoms of hypoglycemia. The physician is in attendance at all times during an application of IPT. Patients are always prepared with ready access to a vein in order to receive supplementary intravenous glucose if necessary on account of any idiosyncratic reactions to insulin.
Blood glucose determinations by Accucheck are done three times during IPT treatments: in the fasting state before administration of the insulin; at the glucose nadir at 25 minutes; and after the recovery phase. Typical values here are 70 to 100 mg % for the initial and final readings, and 40 to 50 mg% at the nadir reading.
There are certain conditions that can alter individual reactivity to low blood glucose levels. The entry of glucose across the blood brain-barrier (BBB) and into the brain is mediated by an insulin-independent glucose transport protein (GTP).(49) The rate at which this BBB-GTP operates to transport glucose varies, as it is up-regulated or down-regulated according to blood glucose levels. The system gets up regulated during prolonged and repeated episodes of hypoglycemia,(50) which explains why certain individuals with insulinomas or chronic reactive hypoglycemia may not have symptoms of brain glucopenia at blood glucose concentrations well below 50 mg %. In a like fashion, the BBB-GTP is down-regulated during prolonged periods of hyperglycemia such as can occur with undiagnosed or poorly controlled type II diabetes.(51) When such patients are brought under rapid control with appropriate therapy, because of this down-regulation of the BBB-GTP, they may develop symptoms of brain glycopenia even though the blood glucose level is in the normal range.(52) These factors have to be kept in mind when interpreting the reactions certain individuals may have during their treatments with Insulin Potentiation Therapy
REFERENCES
1. Porte D, Halter JB. The endocrine pancreas and diabetes mellitus. Williams RH (Ed), Textbook of Endocrinology. W.B. Saunders Company, Philadelphia, p. 742-749, 1981.
2. Schnetzler MB, Rubin JB, Pilch PF. Structural requirements for the transmembrane activation of the insulin receptor kinase. J Biol Chem 261:15281-15287, 1986
3. Heidenreich KA, Olefsky JM. Metabolism of insulin receptors. p 63 in Molecular Basis of Insulin Action (M.P. Czech, editor) Plenum Press, New York, 1985
4. Burdett E, Beeler T, Klip A. Distribution of glucose transporters and insulin receptors in the plasma membrane and transverse tubules of skeletal muscle. Arch Biochem Biophys 253:279-286, 1987
5. Kono T. Translocation hypothesis of insulin action on glucose transport. Federation Proc 43:2256-2257, 1984
6. Rosen OM. After insulin binds. Science 273:1452-1457, 1987 7. Holdaway IM, Freisen HG. Hormone binding by human mammary carcinoma. Cancer Research 37:1946-1952, 1977
8. Wong M, Holdaway IM. Insulin binding by normal and neoplastic colon tissue. Int J Cancer 35:335-341, 1985
9. Mountjoy KG, Holdaway IM, Finlay GJ. Insulin receptor regulation in cultured human tumor cells. Cancer Research 43:4537-4542, 1983
10. Shafie S, Brooks SC. Effect of prolactin on growth and the estrogen receptor level of human breast cancer cells [MCF-7]. Cancer Res 37:792-799, 1977
11. Myal Y, Shiu RPC, Bhomick B, Bala B. Receptor binding and growth promoting activity of insulin-like growth factors in human breast cancer cells [T-47D] in culture. Cancer Res 44:5486-5490, 1984
12. Spring-Mills E, Stearns SB, Smith TH, et al. Immunoreactive hormones in human breast tissues. Surgery 94(6):946-950, 1983
13. Pavelik L, Pavelik K, Vuk-Pavlovic S. Human mammary and bronchial carcinomas: in vivo and in vitro secretion of substances immunologically cross-reactive with insulin. Cancer 53(11):2467-2471, 1984
14. Shames JM, Dhurandhar NR, Blackard WG. Insulin-secreting bronchial carcinoid tumor with widespread metastases. Am J Med 44:632-637, 1968
15. Kiang DT, Bauer GE, Kennedy BJ. Immunoassayable insulin in carcinoma of the cervix associated with hypoglycemia. Cancer 31:801-804, 1973
16. Pavelik K, Bolanca M, Vecek N, et al. Carcinomas of the cervix and corpus uteri in humans: stage-dependent blood levels of substance(s) immunologically cross-reactive with insulin. J Natl Cancer Inst 68:891-894, 1982
17. Pavelic K, Popovic M. Insulin and glucagon secretion by renal adenocarcinoma. Cancer 48:98-100, 1981
18. Oleesky S, Bailey I, Samos S, Bilkus D. A fibrosarcoma with hypoglycemia and a high serum insulin level. Lancet 2:378-380, 1962
19. Pavelic K, Odavic M, Pekic B, et al. Correlation of substance(s) immunologically cross-reactive with insulin, glucose and growth hormone in Hodgkin’s lymphoma patients. Cancer Lett 17:81-86, 1982
20. Bar RS, Roth J. Insulin receptor status in disease states of man. Arch Int Med 137:474-481, 1977
21. Pardridge WM. Receptor-mediated peptide transport through the blood brain barrier. Endocrine Reviews 7:314-330, 1986
22. McCall A, Chick W, Ruderman N. Chronic hypoglycemia increases brain glucose transport and glucose metabolism by brain microvessels. Abstract] Diabetes 32:25A, 1983
23. McCall AL, Millington WR, Wurtman RJ. Metabolic fuel and amino acid transport into the brain in experimental diabetes mellitus. Proc Natl Acad Sci USA 79:5406-5410, 1982
24. DeFronzo RA, Hendler R, Christensen N. Stimulation of counterregulatory hormonal responses in diabetic man by a fall ~n glucose concentration. Diabetes 29:125-131, 1980
25. Pardridge WM, Eisenberg J, Yang J. Human blood-brain barrier insulin receptor. J Neurochem 44:1771-1778, 1985
26. Frank HJL, Jankovic-Vokes T, Pardridge WM, Morris WL. Enhanced insulin binding to blood-brain barrier in vivo and to brain microvessels in vitro in newborn rabbits. Diabetes 34:728-733, 1985
27. Ayre SG. New approaches to the delivery of drugs to the brain. Med Hypoth 29:283-291, 1989
28. Poznansky MJ, Singh R, Singh B, Fantus G. Insulin: Carrier potential for enzyme and drug therapy. Science 223:1304-1306, 1984
29. Yoshimasa S, et al. A New approach to the detection of autoantibodies against insulin receptors that inhibit the internalization of insulin into human cells.Diabetes 33:1051-1054, 1984
30. Gasparro FP, et al. Receptor-mediated photo-cytotoxicity: synthesis of a photoactivatable psoralen derivative conjugated ~o insulin. Biochem Biophys Res Comm 141:502-509, 1986
31. Pardridge WM. Strategies for the delivery of drugs through the blood-brain barrier. p 305-313 in Annual Reports in Medicinal Chemistry-20, Academic Press, 1985
32. Ayre SG, Skaletski B, and Mosnaim AD. The blood-brain barrier passage of azidothymidine: effect of insulin. Res Commun Chem Pathol Pharmacol 63(1):45-52, 1989
33. Cullen JK, Yee D, Sly WS, et al. Insulin-like growth factor receptor expression and function in human breast cancer. Cancer Res 50:48-53, 1990
34. Papa V, Pezzino V, Constantino A, et al. Elevated insulin receptor content in human breast cancer. J Clin Invest 86:1503-1510, 1990
35. Alabaster O, Vonderhaar BK, Shafie SM. Metabolic modification by insulin enhances methotrexate cytotoxicity in MCF-7 human breast cancer cells. Eur J Cancer Clin Oncol 17:1223-1228, 1981
36. Schilsky RL, Bailey BD, Chabner BA. Characteristics of membrane transport of methotrexate by cultured human breast cancer cells. Biochem Pharmacol 30:1537-1542, 1981
37. Eibl H. Phospholipids as functional constituents of biomembranes. Angew Chem Int Ed Engl 23:257-271, 1984
38. Jeffcoat R and Jame AT. The regulation of desaturation and elongation of fatty acids in mammals. Numa S. (Ed), Fatty Acid Metabolism and its Regulation. Elsevier Science Publishers BN. p.85-112, 1984.
39. Jeffcoat R. The biosynthesis of unsaturated fatty acids and its control in mammalian liver. Essays Biochem 15:1-36, 1979.
40. Zapf J, Froesch ER. Insulin-like growth factors/somatomedins: structure, secretion, biological actions and physiological role. Hormone Res 24:121-130, 1986
41. Lippman ME, Dickson RB, Kasid A, et al. Autocrine and paracrine growth regulation of human breast cancer. J Steroid Biochem 24:147-154, 1986
42. Hilf R. The actions of insulin as a hormonal factor in breast cancer. In: Pike MC, Siiteri PK, Welsch CW, eds. Hormones and Breast Cancer, Cold Spring Harbor Laboratory, 1981, 317-337.
43. King GL, Kahn CR, Rechler MM, Nissley SP. Direct demonstration for separate receptors for growth and metabolic activities of insulin and multiplication-stimulating activity (an insulin-like growth factor) using antibodies to the insulin receptor. J Clin Invest 66:130-140, 1980
44. Cryer PE. Glucose homeostasis and hypoglycemia. Williams RH (Ed), Textbook of Endocrinology. W.B. Saunders Company, Philadelphia, p. 989-1017, 1985.
45. Gerich J, Davis J, Lorenzi M, et al. Hormonal mechanisms of recovery from insulin-induced hypoglycemia in man. Am J Physio1236:E380-E385, 1979.
46. Cryer PE, Tse TF, Clutter WE, and Shah SD. Roles of glucagon and epinephrine in hypoglycemic and nonhypoglycemic glucose counterregulation in humans. Am J Physiol 247:E198-E205, 1984.
47. Rizza RA, Cryer PE, Gerich JE. Role of glucagon, catecholamines, and growth hormone in human glucose counterregulation. J Clin Invest 64:62-71, 1979
48. Clarke WL, Santiago JV, Thomas L, et al. Adrenergic mechanisms in recovery from hypoglycemia in man: adrenergic blockade. Am J Physiol 236:E147-E152, 1979.
49. Pardridge WM. Receptor-mediated peptide transport through the blood-brain. Endocrine Reviews 7:314-330, 1986
50. McCall A, Chick W, Ruderman N. Chronic hypoglycemia increases brain glucose transport and glucose metabolism by brain microvessels. (Abstract) Diabetes 32:25A, 1983
51. McCall A, Millington WR, Wurtman RJ. Metabolic fuel and amino acid transport into the brain in experimental diabetes mellitus. Proc Natl Acad Sci USA 79:5406-5410, 1982
52. DeFronzo RA, Hendler R, Christensen N. Stimulation of counterregulatory hormonal responses in diabetic man by a fall in glucose concentration. Diabetes 29:125-131, 1980